Удобную методологию анализа геномных данных разработали в Назарбаев Университете
Исследователи Лаборатории биоинформатики и системной биологии Центра наук о жизни, ЧУ «National Laboratory Astana» (NLA), Назарбаев Университета в сотрудничестве с исследователями Группы вычислительной системной биологии рака Института Кюри (Париж, Франция) разработали улучшенную методологию анализа геномных данных. Методология BiODICA облегчает проведение анализа с помощью Метода Независимых Компонент (МНК) и оценку результатов, а также позволяет исследователям со всего мира сосредоточиться на интерпретации результатов анализа данных и выводе гипотез исследования.
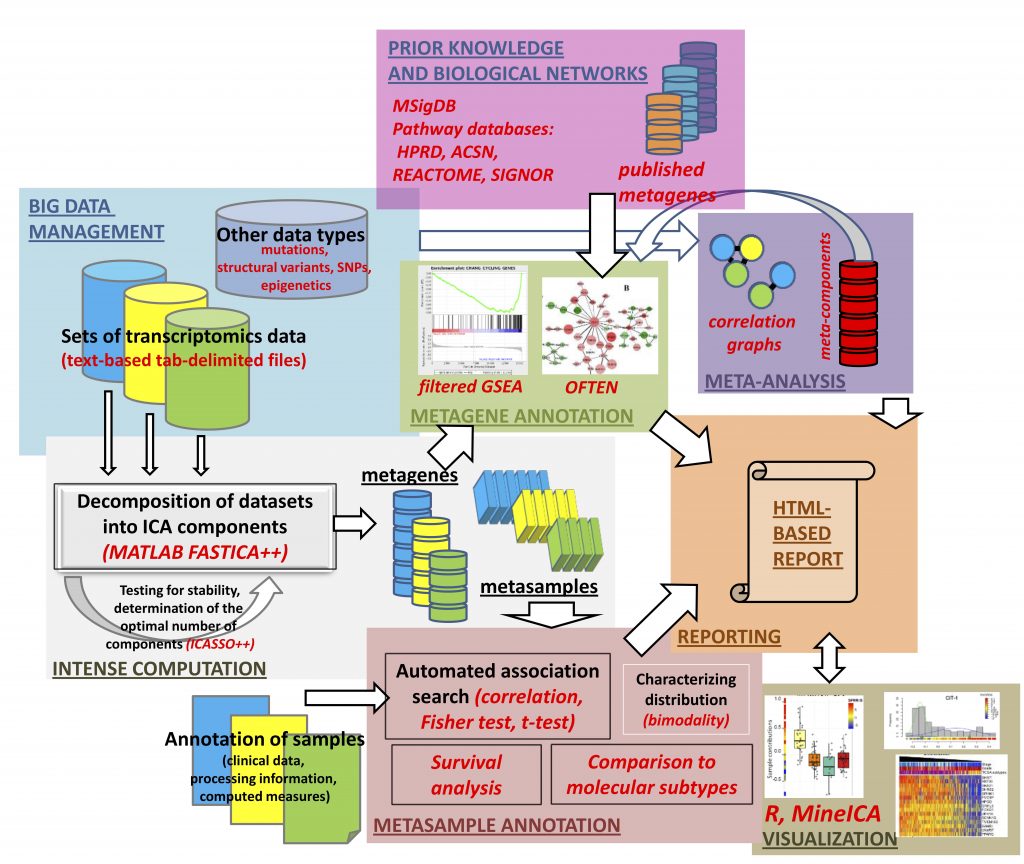
Дело в том, что при геномных исследованиях по изучению рака, в результате работы с высокопроизводительными геномными платформами, генерируется огромное количество молекулярных данных, которые, как правило, сложно анализировать, и интерпретировать из-за их многомерности и множества других факторов, влияющих на молекулярные профили раковых клеток и тканей. Так, одним из современных методов анализа многомерных геномных данных является МНК. Однако и у него есть свои недостатки, связанные с высокими требованиями к вычислительным ресурсам, зависимость от программных сред разработки (Matlab, R Bioconductor), определением оптимального числа необходимых компонент, отсутствием интуитивного способа сравнения независимых компонент и отсутствием удобного программного интерфейса.
Напомним, ранее исследователи Лаборатории биоинформатики и системной биологии уже запустили первую в Казахстане высокопроизводительную биоинформатическую вычислительную платформу для «больших геномных данных» и решения задач в области биоинформатики Q-Symphony или “Qazaq symphony of bioinformatics”. Разработанная методология анализа геномных данных BiODICA совместима с любой из известных биоинформатических вычислительных платформ и более того, может быть реализована на обычном персональном компьютере.
По мнению исследователей NLA первые результаты применения разработанной методологии показали ее преимущество в сравнении с классическими методами (Метод главных компонент, кластеризация) в отношении биологической интерпретации извлеченных сигналов, где эти компоненты могут отражать как биологические факторы (например, пролиферация или присутствие различных типов клеток в опухолевой ткани и ее микроокружении), так и технические факторы (например, “batch-эффекты” или GC-содержание), влияющих на экспрессию (активность) генов.
— Последние десять лет совместно с исследовательскими группами доктора Андрея Зиновьева (A.Zinovyev) и Эммануэля Барилло (E.Barillot) из Института Кюри (Париж, Франция) мы изучали большие наборы онко-данных и работали над созданием эффективной методологии анализа и интерпретации данных с использованием МНК. Нам удалось доработать и оптимизировать данную методологию. Мы сравнили результаты, полученные в сравнении с рядом других применяемых методов: методом главных компонент, методом неотрицательной матричной факторизации и классическим МНК-анализом. Наш биоинформатический подход с использованием МНК подтвердил надежность и эффективность разработанной методологии для анализа больших наборов раковых транскриптомов и может быть полезным для широкого круга исследователей, заинтересованных в анализе «больших геномных» данных, — говорит Улыкбек Каиров, ведущий научный сотрудник, руководитель Лаборатории биоинформатики и системной биологии, Центра Наук о Жизни, ЧУ «National Laboratory Astana».
Методология BiODICA состоит из нескольких основных модулей:
— автоматизации деконволюции больших “омиксных” наборов данных с оптимизацией параметров деконволюции;
— интерпретация результатов применения деконволюции путем аннотирования компонент с использованием различных методов;
— сравнения результатов деконволюции независимых наборов данных для изучения воспроизводимых сигналов, универсальных и специфических для конкретного типа / подтипа рака / заболевания.
В дальнейшем исследователи NLA планируют интегрировать различные наборы геномных данных для улучшения методологии анализа, а также продолжат изучать молекулярные сигналы в опухолевых тканях.
Научные статьи по этому исследованию:
1) Blind source separation methods for deconvolution of complex signals in cancer biology, Zinovyev, A., Kairov, U. et al. Biochem Biophys Res Commun., 2013;
2) Determining the optimal number of independent components for reproducible transcriptomic data analysis, Kairov U. et al. BMC Genomics, 2017;
3) Application of Independent Component Analysis to Tumor Transcriptomes Reveals Specific and Reproducible Immune-Related Signals, Czerwinska, U. et al. Lect. Notes in Comp. Sci., 2018;
4) Assessing reproducibility of matrix factorization methods in independent transcriptomes, Cantini, L., Kairov U., et al. Bioinformatics, 2019;