An advanced methodology for genomic analysis developed at Nazarbayev University
Researchers of the Laboratory of Bioinformatics and Systems Biology of the Center for Life Sciences, National Laboratory Astana (NLA), Nazarbayev University in collaboration with scientists of Computational Systems Biology of Cancer Group (Institut Curie, Paris, France) developed an advanced methodology for genomic analysis. With the help of the methodology of Independent Component Analysis (ICA) BiODICA analysis is becoming easier, and this is allowing scientists from all over the world to focus on interpreting their results and to create research hypotheses.
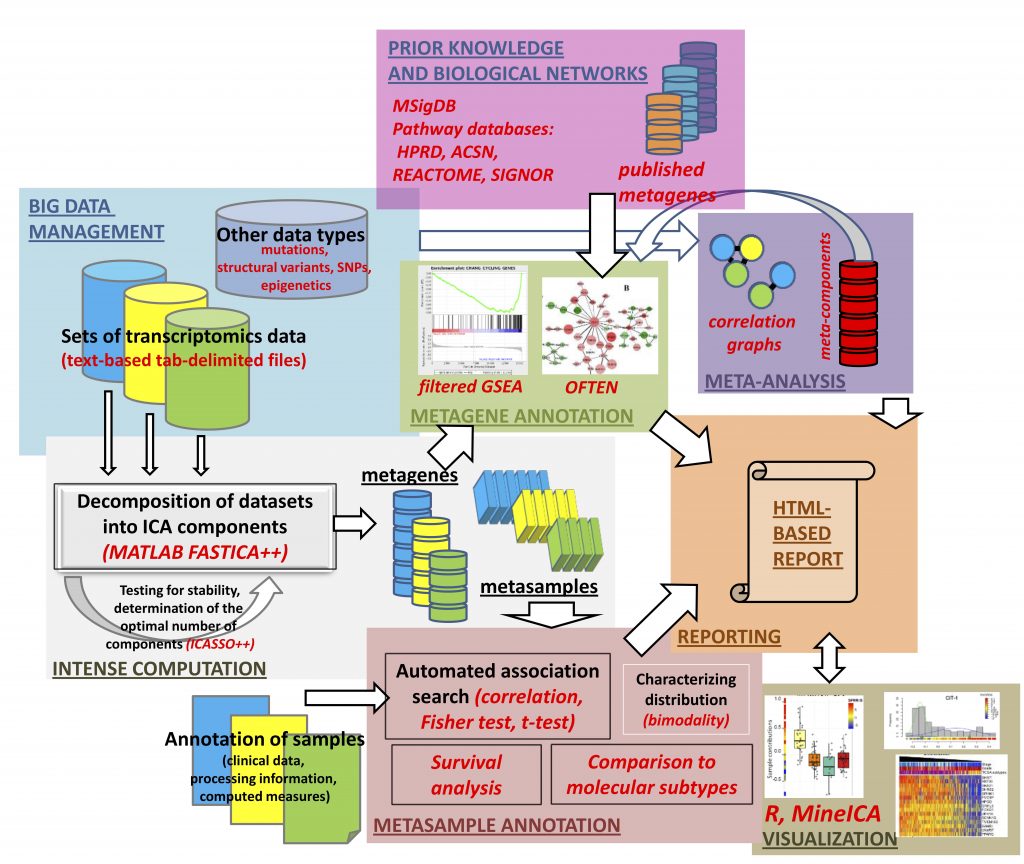
Within genomic research, massive amounts of molecular data are derived from high-throughput genomic platforms creating difficulties in analysis and interpretation due to the intrinsic multi-dimensionality and many other factors linked to molecular profiles of tumor cells and tissues. Thus, one of the modern analysis methods of multidimensional genomic data is ICA. However, using ICA also has its own disadvantages associated with high demands on computational resources, dependence on software development environments (Matlab, R Bioconductor), the definition of the optimal number of necessary components, the lack of an intuitive way to compare independent components, and the absence of a user-friendly software interface.
It should be noted that recently researchers of the Laboratory of Bioinformatics and Systems Biology launched the first high-performance bioinformatics computational platform in Kazakhstan Q-Symphony or “Qazaq Symphony of Bioinformatics” for dealing with the complexity of “big genomic data” and solving problems in the field of bioinformatics. The methodology developed for genomic analysis BIODICA is compatible with any of the already existing bioinformatics computational platforms and even can be used on a regular personal computer.
According to NLA researchers, the first results produced by using BiODICA have shown advantages when compared to using classic methods such as hierarchical clustering, Principal Component Analysis. Specifically, with respect to biological interpretability of the resulting components, where these components might reflect both biological factors such as proliferation or presence of different cell types in the tumoral microenvironment, or technical factors such as batch effects or GC-content affecting gene expression.
– Over the past ten years, we have worked with the research teams of Dr. Andrei Zinovyev and Emmanuel Barillot of the Curie Institute (Paris, France) to study large sets of cancer data and to develop an effective methodology for analyzing and interpreting data using ICA. We were able to refine and optimize this methodology. We studied the results obtained in comparison with a number of other applied methods: Principal Component Analysis, Non-negative Matrix Factorization method, and classical ICA analysis. Our bioinformatics approach with using the ICA method confirmed a robustness and usefulness of the developed methodology for analysis of big transcriptomics datasets and would be helpful for a broad biomedical community interested in analysis of cancer data, – said Dr. Ulykbek Kairov, Leading researcher, Head of the Laboratory of Bioinformatics and Systems Biology, Center for Life Sciences, NLA.
BiODICA implemented in Java language with several modules for:
(1) automating deconvolution of large omics datasets with optimization of deconvolution parameters;
(2) helping in interpretation of the results of deconvolution application by automated annotation of the components using the best practices;
(3) comparing the results of deconvolution of independent datasets for distinguishing reproducible signals, universal and specific for particular cancer/disease type or subtype.
Further on, NLA researchers plan to integrate various genomic data sets to improve the analysis methodology and will continue to study the molecular signals in tumor tissues.
List of articles on this research:
1) Blind source separation methods for deconvolution of complex signals in cancer biology, Zinovyev, A., Kairov, U. et al. Biochem Biophys Res Commun., 2013;
2) Determining the optimal number of independent components for reproducible transcriptomic data analysis, Kairov U. et al. BMC Genomics, 2017;
3) Application of Independent Component Analysis to Tumor Transcriptomes Reveals Specific and Reproducible Immune-Related Signals, Czerwinska, U. et al. Lect. Notes in Comp. Sci., 2018;
4) Assessing reproducibility of matrix factorization methods in independent transcriptomes, Cantini, L., Kairov U., et al. Bioinformatics, 2019;