Назарбаев Университетінде геномдық деректерді талдаудың ыңғайлы әдіснамасын әзірледі
Назарбаев Университетінің “National Laboratory Astana” ЖМ (NLA) Өмір туралы ғылымдар орталығының Биоинформатика және жүйелік биология зертханасының зерттеушілері Кюри институтының (Париж, Франция) обыр есептеуіш жүйелік биологиясы тобының зерттеушілерімен ынтымақтастықта геномдық деректерді талдаудың жақсартылған әдіснамасын әзірледі. BiODICA әдіснамасы тәуелсіз компонент әдісімен талдау жүргізуді және нәтижелерін бағалауды жеңілдетеді, сонымен қатар бүкіл әлем бойынша зерттеушілерге деректерді талдау нәтижелерін түсіндіруге және зерттеу гипотезаларын жасауға ден қоюға мүмкіндік береді.
Себебі обырды зерттеу бойынша геномдық зерттеулер кезінде жоғары өнімді геномдық платформалармен жұмыс істеу нәтижесінде көптеген молекулалық деректер жасалады. Әдетте, олардың санқырлылығы мен обыр жасушалары және тіндерінің молекулалық бейіндеріне әсер ететін басқа да факторлардың көптігінен, оларды талдау және түсіндіру қиын. Осылайша, санқырлы геномдық деректерді талдаудың қазіргі заманғы әдістерінің бірі ТМК болып табылады. Алайда, есептеуіш ресурстарға қойылатын жоғары талаптарға байланысты оның да өз кемшіліктері бар. Атап айтқанда, әзірлеудің бағдарламалық ортасына тәуелділігі (Matlab, R Bioconductor), қажетті компоненттердің оңтайлы санын анықтау, тәуелсіз компоненттерді салыстырудың интуитивті тәсілінің болмауы және ыңғайлы бағдарламалық интерфейстің болмауы.
Естеріңізге сала кетейік, бұған дейін Биоинформатика және жүйелік биология зертханасының зерттеушілері «үлкен геномдық деректер» және Q-Symphony биоинформатика немесе “Qazaq symphony of Bioinformatics” саласындағы міндеттерді шешу үшін Қазақстанда алғашқы жоғары өнімді биоинформатикалық есептеу платформасын іске қосты. BiODICA геномдық деректерді талдаудың әзірленген әдіснамасы белгілі биоинформатикалық есептеуіш платформалардың кез келгенімен үйлесімді және сонымен қатар, әдеттегі дербес компьютерде іске асырылуы мүмкін.
NLA зерттеушілерінің пікірінше, әзірленген әдіснаманы қолданудың алғашқы нәтижелері алынған сигналдарды биологиялық интерпретациялауға қатысты классикалық әдістермен (басты компонент әдісі, кластерлеу) салыстырғанда оның артықшылығын көрсетті, онда бұл компоненттер биологиялық факторлар (мысалы, ісік тініндегі және оның микро ортасындағы жасушалардың әртүрлі типтерінің пролиферациясы немесе олардың болуы), сондай-ақ гендердің экспрессиясына (белсенділігіне) әсер ететін техникалық факторлар (мысалы, “batch-әсерлер” немесе GC-мазмұны).
– Соңғы он жыл бойы Кюри институтының (Париж, Франция) докторы Андрей Зиновьев (A.Zinovyev) және Эммануэль Бариллоның (E. Barillot) зерттеу топтарымен бірлесіп, біз онко-деректердің үлкен жиынтығын зерттей отырып, МНК-ні пайдаланып, деректерді талдау мен түсіндірудің тиімді әдіснамасын жасадық. Біз осы әдіснаманы пысықтап, оңтайландырдық. Біз басқа да қолданылатын әдістермен: басты компонент әдісімен, теріс емес матрицалық факторизация әдісімен және классикалық МНК-талдаумен салыстырғанда алынған нәтижелерді салыстырдық. Біздің МНК пайдаланылатын биоинформатикалық тәсіліміз обыр транскриптомдарының үлкен жиынтығын талдау үшін әзірленген әдіснаманың сенімділігі мен тиімділігін растады және “үлкен геномдық” деректерді талдауға мүдделі зерттеушілердің көбісіне пайдалы болуы мүмкін, – дейді жетекші ғылыми қызметкер, “National Laboratory Astana” ЖМ, Өмір туралы ғылым орталығы, Биоинформатика және жүйелік биология зертханасының жетекшісі Ұлықбек Қайыров.
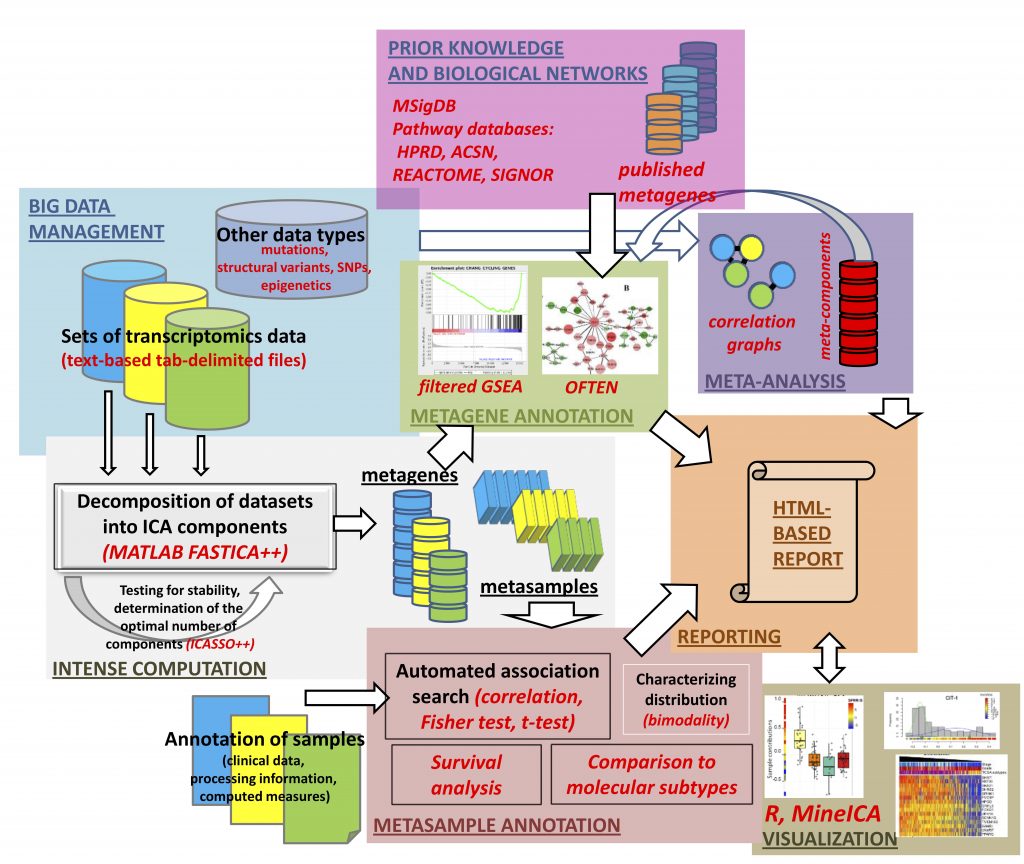
BiODICA әдіснамасы бірнеше негізгі модульдерден тұрады:
– деконволюция параметрлерін оңтайландырумен үлкен “омикс” деректер жиынтығының деконволюциясын автоматтандыру;
– әр түрлі әдістерді қолдана отырып аннотациялау арқылы деконволюцияны қолдану нәтижелерін түсіндіру;
– обырдың / аурудың нақты түріне / шағын түріне арналған әмбебап және ерекше, жаңғыртылатын сигналдарды зерттеу үшін тәуелсіз деректер жиынтығының деконволюциясының нәтижелерін салыстыру.
Бұдан әрі NLA зерттеушілері талдау әдіснамасын жақсарту үшін геномдық деректердің әртүрлі жиынтығын біріктіруді жоспарлап отыр, сондай-ақ ісік тіндеріндегі молекулалық сигналдарды зерттеуді жалғастырады.
Бұл зерттеу бойынша ғылыми мақалалар:
1) Blind source separation methods for deconvolution of complex signals in cancer biology, Zinovyev, A., Kairov, U. et al. Biochem Biophys Res Commun., 2013;
2) Determining the optimal number of independent components for reproducible transcriptomic data analysis, Kairov U. et al. BMC Genomics, 2017;
3) Application of Independent Component Analysis to Tumor Transcriptomes Reveals Specific and Reproducible Immune-Related Signals, Czerwinska, U. et al. Lect. Notes in Comp. Sci., 2018;
4) Assessing reproducibility of matrix factorization methods in independent transcriptomes, Cantini, L., Kairov U., et al. Bioinformatics, 2019;